
标准物质一站式采购平台


新闻
- 产品
- 帖子
- 新闻
金属混标查询
在线客服
标准物质一站式采购平台
19 种性激素均为环戊烷多氢菲衍生物,其中有4 组共12 种化合物互为同分异构体,甚至差向异构体,实验需选用合适的色谱柱,以实现上述化合物的基线分离,避免具有相同m/z的母离子和子离子相互干扰。为此,实验分别考察Torus 2-PIC(3.0 mm×100 mm,1.7 μm)、HSS C18 SB(2.1 mm×150 mm,1.8 μm)、BEH(3.0 mm×100 mm,1.8 μm)3 种色谱柱对分离效果的影响。结果表明,选用HSS C18 SB色谱柱和BEH色谱柱均无法实现11α-羟孕酮和11β-羟孕酮、雄酮和表雄酮等差向异构体的基线分离;选用Torus 2-PIC色谱柱时,各目标化合物分离效果最好,可能由于该色谱柱填料提供的偶极-偶极、电子转移、π-π等多重作用可有效识别出目标物空间结构的微小差异。但选用Torus 2-PIC色谱柱时,雌激素色谱峰略有拖尾,仍需进一步优化其他色谱条件。
超临界CO2的洗脱能力会随色谱柱温度和系统背压的改变而变化,当柱温升高、背压减小时,流动相密度变小,洗脱能力降低,化合物保留时间延长。实验分别考察不同柱温(25、35、45、55 ℃)、不同背压(12.07、13.10、13.79、14.48 MPa)对分离效果的影响。结果表明,化合物的分离效果并未随背压的改变而出现明显变化;柱温升高时,分离趋势增加,当柱温超过45 ℃后,分离度反而减小,可能因为升高柱温加剧了分子的热运动,从而改变了化合物在色谱柱中的保留特性。综合考虑分离度、系统压力极限等因素,选取45 ℃色谱柱温度、13.79 MPa系统背压为测定条件。
使用UPCC时,向流动相中引入适量的助溶剂可有效改善超临界CO2的溶解能力和洗脱能力,常用助溶剂的洗脱能力由强到弱依次为甲醇、乙醇、异丙醇、乙腈,使用甲醇作为助溶剂时,不仅可以加快分析速度,还能获得更为尖锐的色谱峰形。性激素的分子结构中含有羰基、酚羟基等官能团,这些极性基团极易与色谱柱填料表面未键合的硅醇羟基产生氢键作用,从而引起色谱峰拖尾。Quanson等的研究显示,当流动相的pH值较低时,可以改善色谱峰的拖尾现象。因此,实验在甲醇中添加体积分数0.1%甲酸。结果表明,使用0.1%甲酸-甲醇溶液作为助溶剂时,雌激素的色谱峰形更加对称和尖锐。
实验在优化的色谱条件下对19 种性激素混合标准溶液(20 μg/L)进行测定,其提取离子流色谱图见图1。经计算,图1中个别性激素色谱峰的理论塔板数高达105,由此表明,UPCC具有出色的分离效率。
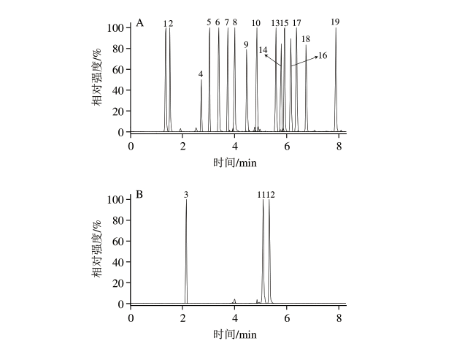
图1 19 种性激素混合标准溶液(20 μg/L)的提取离子流色谱图
将稀释后的标准品储备溶液直接注入离子源,分别在正、负两种电离模式下优化质谱条件。结果表明,在正离子模式下,雄激素和孕激素分子结构中的羰基、共轭双键等结构更易于获得氢离子,从而生成较高丰度的[M+H]+母离子;雄酮、表雄酮、脱氢表雄酮在获得氢离子后,经过开环、重排等复杂的反应,使[M+HH2O]+母离子丰度高于其他加合物形式;在负离子模式下,雌激素则更易于失去酚羟基上的氢,生成较高丰度的[M-H]-母离子。确定母离子后,开启IntelliStart自动调谐功能,采用自动调谐所得的多反应监测参数。
在ESI源的金属毛细管中,超临界CO2因失去压力控制而转化为气态,从而失去溶解能力。为使分析物能够顺利传输至离子源并发生电离,需在色谱柱后端引入适宜的补偿液。补偿液不参与色谱分离,但其组成和流速对化合物的色谱峰形和离子化效率具有重要影响。Sun Ping等的研究表明,使用甲酸酸化后的质子性溶剂作为补偿液更有利于提高分析物在正离子模式下的质谱响应,且流动相pH值至少低于样品pKa值2 个单位;使用碱化后的非质子性溶剂作为补偿液更有利于待测物[M-H]-分子离子峰的形成,且流动相pH值至少高于样品pKa值2 个单位。因此,实验分别考察不同体积分数97%甲醇溶液(含体积分数0.1%、0.2%、0.3%甲酸)和不同体积分数98%乙腈溶液(含体积分数0.1%、0.2%、0.3%氨水)作为补偿液,以及补偿液在不同流速(0.1、0.2、0.3、0.4 mL/min)时对测定结果的影响。结果表明,选用97%甲醇溶液(含体积分数0.2%甲酸)和98%乙腈溶液(含体积分数0.2%氨水)分别作为正、负离子模式下的补偿液,可获得较好的色谱峰形和响应强度;补偿液流速为0.3 mL/min时,各性激素响应值最高(图2a);继续增加流速,则系统压力超过设定值,迫使背压阀开启排放功能,分流了部分性激素,使质谱响应值降低。
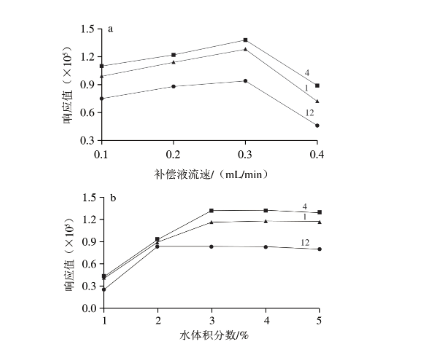
图2 补偿液流速(a)和补偿液中纯水体积分数(b)对3 种性激素峰高的影响
此外,在补偿液中加入适量纯水,可改变离子源中的气氛,有助于提高化合物的质谱响应。通过比较补偿液中添加不同体积分数(1%、2%、3%、4%、5%、6%)纯水时化合物的峰高(图2b),可知正、负离子模式下纯水的最佳体积分数分别为3%和2%;当超过5%后,系统压力波动超过仪器告警阈值,仪器始终无法进入就绪状态,可能因为此时流动相管路内难以形成稳定的超临界流体。
性激素极性较弱,可选用甲醇、乙腈、丙酮等作为提取液。实验参考饶雅琨等的研究,选用甲醇提取固体样品中的性激素。李双等研究表明,在蛋白粉中加入适量纯水,可使样品产生溶胀,提高提取液的穿透性;酸性环境能有效降低雌激素在水中的溶解度,从而提高有机溶剂的提取效率。因此,实验以蛋白粉阴性加标样品(20 μg/kg)为对象,考察直接提取和加水润湿后提取、提取液中含不同体积分数(0%、1%、2%,图3a)甲酸,以及不同超声提取时间(5、10、15、20 min,图3b)对回收率的影响。结果显示,将样品润湿后,以1%甲酸-甲醇溶液超声提取15 min时各性激素回收率最高。

图3 不同体积分数甲酸(a)和超声提取时间(b)对19 种性激素回收率的影响
一些研究采用凝胶制备色谱法提取和净化油状样品中的性激素,但该方法效率低下,成本高昂且污染严重。因此,实验改进了黄百芬等的研究,先将样品溶解于正己烷中,再用硅胶粉末吸附溶液中的性激素,甘油酯、VE等非极性杂质则随正己烷一同弃去,最后用甲醇洗脱硅胶上吸附的性激素。该方法可除去样品中的大部分杂质,降低后续净化的难度,并且操作简单、成本低廉。实验以19 种性激素的正己烷溶液(10.0 μg/L)为模拟物进行验证,结果显示,经硅胶粉末吸附后的正己烷溶液中未检出性激素,而甲醇洗脱液中各性激素质量浓度均不小于9.7 μg/L。
以上样品提取液中仍含有大量杂质,需采取有效的净化手段进一步减小基质干扰。为提高分析通量,实验采用通过式固相萃取法净化样品提取液。张再永等的研究表明,PRiME HLB通过式反相固相萃取柱能有效保留甘油酯、磷脂、蛋白质等化合物,且使用前无需活化和平衡,可极大的提高工作效率。甲醇、乙腈均是反相固相萃取法常用的洗脱液,乙腈的洗脱能力更强,有利于减少试剂消耗并缩短浓缩时间。因此,实验以PRiME HLB固相萃取柱为净化柱,乙腈为洗脱液净化样品提取液。通过比较不同洗脱体积(0、1、2、3、4、5 mL)下各性激素的回收率可知,最佳洗脱体积为4 mL;超过4 mL后,回收率不再变化,但一级质谱扫描图谱中杂质峰簇明显增多。因此,实验最终采用4 mL乙腈进行洗脱。
实验配制并测定了系列基质匹配标准溶液和系列空白溶剂标准溶液,基质效应=基质匹配标准曲线斜率/空白溶剂标准曲线斜率-1。结果表明,3 种样品的基质效应均在-0.51~0.20范围内,其中蛋白粉基质对雌激素具有较强的基质抑制效应,可能是在离子化过程中,溶液中残留的蛋白质、多肽等强极性杂质与雌激素共同竞争液滴表面,导致雌激素离子化效率降低;VE软胶囊基质中,各性激素的基质效应普遍较弱,这与张虹艳等的研究结论一致。为减小基质效应对定量分析准确性的影响,实验采用空白基质匹配标准溶液绘制标准曲线。
以阴性样品提取液配制各性激素质量浓度均为0.10~100 μg/L的基质匹配标准溶液,采用本方法进行分析,以目标物定量离子峰面积为纵坐标,对应质量浓度为横坐标绘制标准曲线。结果表明,19 种性激素在各自的质量浓度范围内线性关系良好,相关系数(R2)不小于0.997 1。再以各目标化合物3 倍信噪比(RSN=3)确定LOD,以10 倍信噪比(RSN=10)确定LOQ。各性激素的LOD和LOQ分别为0.03~0.85 μg/kg和0.10~2.5 μg/kg,与Toit[30]、Quanson等报道的数值水平相当,优于李双、饶雅琨、罗文静等的报道。
以阴性样品进行加标回收实验,加标量分别为0.5、2 mg/kg和10 mg/kg,每个加标水平制备3 份,按1.3.1和1.3.2节方法处理后进行测定,得到样品的平均回收率(n=9)为77.8%~111.7%,相对标准偏差(relative standard deviation,RSD)为1.1%~8.4%。与李双、饶雅琨、罗文静等的研究相比,回收率和精密度均能满足保健食品中性激素非法添加检测的要求。
按照建立的方法对2 份VE软胶囊样品、2 份胶原蛋白粉样品和4 份蛋白粉样品进行测定。其中1 份胶原蛋白粉样品中检出孕酮和雌三醇,含量分别为1.42、0.88 mg/kg;1 份蛋白粉样品检出诺龙、睾酮和雌三醇,含量分别为11.05、2.21 mg/kg和0.40 mg/kg;1 份蛋白粉样品检出诺龙、睾酮和甲睾酮(图4),含量分别为7.39、1.53 mg/kg和0.59 mg/kg,其他样品均未检出这19 种性激素。蛋白粉类样品的总体检出率为50%,与李双等的研究基本一致。
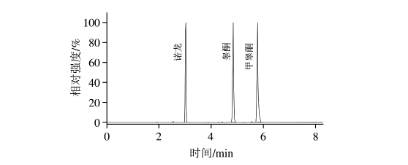
图4 阳性蛋白粉样品(6号样品)的总离子流色谱图
建立UPCC-串联质谱测定保健食品中19 种性激素含量的方法,采用优化的样品前处理方式,有效减小了基质干扰,并提高了检测灵敏度。方法快速准确、灵敏度高、专属性好、绿色环保,可在8 min内有效分离包括2 组4 种差向异构体在内的4 组共12 种同分异构体。采用建立的方法对实际样品进行检测,结果能满足一般化学分析的要求,对于保健食品中性激素的测定和暴露风险评估提供了可靠的技术支持。
相关链接:环戊烷多氢菲,11β-羟孕酮,乙醇,异丙醇,乙腈,甘油酯,甲醇溶液,北纳生物
本文章来源于——《食品科学网》,如有版权问题,请与本网联系



随着科研领域的不断拓展和深入,极端环境微生物与肿瘤微环境的研究日益受到学术界的关注。为了更好地探讨这两个领域的最新进展和前沿技术,我们特别策划了4月份的专题研讨会,为广大科研工作者带来一场学术盛宴。
了解更多> >
通话对您免费,请放心接听
温馨提示:
1.手机直接输入,座机前请加区号 如13164239859,010-58103778
2.我们将根据您提供的电话号码,立即回电,请注意接听
3.因为您是被叫方,通话对您免费,请放心接听



登录后才可以评论