
标准物质一站式采购平台


新闻
- 产品
- 帖子
- 新闻
金属混标查询
在线客服
标准物质一站式采购平台
目前,氟虫腈的检测方法主要有高效液相色谱(high performance liquid chromatography,HPLC)法、高效液相色谱-串联质谱(high performance liquid chromatography-tandem mass spectrometry,HPLCMS/MS)法、气相色谱法以及气相色谱-质谱(gas chromatography-mass spectrometry,GC-MS)联用法,这些方法各有优缺点。HPLC-MS/MS、GC-MS具有较高的灵敏度,应用范围较广,但检测成本相对较高,需要较为贵重的精密仪器,操作人员技能要求较高。HPLC检测成本低,操作较为简便,但灵敏度不高。因此,开发灵敏度高、选择性强、干扰小的氟虫腈残留检测方法具有重大意义。
高效液相色谱-荧光检测(high performance liquid chromatography-fluorescence detection,HPLC-FLD)灵敏、稳定、干扰小,因此被广泛应用于痕量和微量检测。HPLC-FLD检测的灵敏度主要取决于荧光衍生试剂的性质,高荧光强度就意味着高灵敏度。一个优良的荧光衍生试剂必需具备一个高强度的发色基团和一个高效率的衍生基团。在荧光衍生试剂的分子结构中,发色团是最为重要的部分,可以说该基团决定了HPLC-FLD的检测灵敏度,因此发色团的设计合成又是荧光衍生试剂设计合成的核心。此外,衍生化反应基团是荧光衍生试剂另一个重要的必需结构,是连接待测物与荧光体的“手臂”,这个基团决定了衍生化反应效率的高低。
香豆素类化合物具有良好的光物理和光化学性质,对环境有极强的敏感性,同时具有较大的Stokes位移,因此在荧光检测中具有很好的应用前景。该类物质分子结构中的取代基位置和类型对其荧光性质有较大的影响,不同位置的供电子基团和吸电子基团能在其结构上建立“推、拉”电子体系,可减弱或增强该体系,进而有效地控制荧光强度;同时,其结构中的供、吸电子基团容易被修饰,从而使其荧光波长在大且宽的范围之内得到更好的拓展。因此,香豆素类化合物作为荧光衍生试剂还具有很大的改造和提升空间。
研究表明,氟虫腈分子结构中吡唑环上3位腈基、4位三氟甲基亚砜基及5位游离的氨基均可被修饰和改造,可利用这一结构特点设计和合成氟虫腈柱前荧光衍生试剂,从而建立氟虫腈HPLC-FLD检测方法。相比较而言,吡唑环上的5位氨基更易被修饰,因此针对该基团进行特异性反应生成较强荧光强度的氟虫腈衍生物是可行的。
以4-(二乙氨基)水杨醛为母体,合成以羧酸基团为衍生化反应基团的香豆素类荧光衍生试剂——7-(二乙氨基)-2-氧-2H-色酮-3-羧酸(记为L1),以L1与氟虫腈进行衍生化反应,生成柱前荧光衍生产物N-(3-氰基-1-(2,6-二氯-4-(三氟甲基)苯基)-4-((三氟甲基)亚磺酰基)-1H-吡唑-5-基)-7-(二乙氨基)-2-氧-2H-色酮-3-甲酰胺(记为SF1),在此基础上建立高灵敏度的氟虫腈HPLC-FLD检测方法。本实验所合成的结构新颖的香豆素类荧光衍生试剂L1具有高荧光强度,因此在HPLC-FLD检测中具有较高的应用价值;基于羧基和氨基建立的柱前衍生化反应方法,还可用于其他具有氨基基团的待测目标物的检测,因此,本实验检测设计思路和研究方案对HPLCFLD方法研究具有一定的指导意义。
4-(二乙氨基)水杨醛(纯度≥98%) 天津希恩思生化科技有限公司;丙二酸二乙酯(纯度≥99%) 上海毕得医药科技有限公司;乙醇、氢氧化钠、二氯甲烷(dichloromethane,DCM)、N,N-二甲基甲酰胺(N,N-dimethylformamide,DMF)、盐酸(均为分析纯) 西陇科学股份有限公司;氟虫腈(纯度≥98%)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride,EDC,纯度≥99%) 萨恩化学技术(上海)有限公司;哌啶(分析纯)、四氢呋喃(tetrahydrofuran,THF,化学纯) 国药集团化学试剂有限公司;4-二甲氨基吡啶(4-dimethylaminopyridine,DMAP,分析纯)、N,N-二环己基碳酰亚(dicyclohexyl carbodiimide,DCC,分析纯) 上海阿拉丁生化科技股份有限公司;六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(benzotriazole-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate,PyBOP,分析纯)、三乙胺(triethylamine,TEA,分析纯) 西宝生物科技(上海)股份有限公司;二甲基亚砜(dimethyl sulfoxide,DMSO,纯度≥99%) 上海索来宝科技有限公司;乙腈(acetonitrile,ACN,色谱纯)美国Tieda试剂有限公司。
400/500MHz核磁共振波谱仪 德国布鲁克科技有限公司;7890A-5975C GC-MS联用仪、1260-G1321B HPLC-FLD仪 安捷伦科技有限公司;SGW X-4B显微数字熔点测定仪 上海仪电科学仪器股份有限公司;DLSB-5/25低温冷却液循环泵 巩义市予华仪器有限责任公司;MR Hei-End恒温加热磁力搅拌器、Precision MLG3旋转蒸发仪 德国海道夫集团;PS-06A数控超声波清洗仪 深圳市恒达康科技有限公司;超纯水制备仪 美国Milli-Q科技有限公司。
在实验室前期合成工作基础上[31],以4-(二乙氨基)水杨醛为原料,在哌啶催化下与丙二酸二乙酯缩合成环,再经酯水解等反应,合成荧光衍生试剂L1,具体合成路线如图1所示。
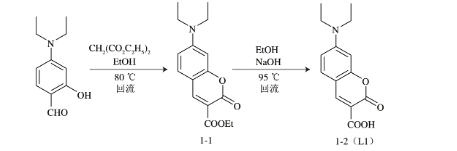
图1 香豆素类荧光衍生试剂(L1)的合成路线
中间体化合物1-1的合成:分别将4-(二乙氨基)水杨醛(9.7 g,1.0 mmol)和丙二酸二乙酯(16.0 g,2.0 mmol)溶于150 mL无水乙醇溶液中,添加10 mL哌啶,将其混合溶液在80 ℃搅拌反应6 h,薄层色谱(thin layer chromatography,TLC)法监测反应进程。待反应结束,溶液冷却至室温,脱溶,硅胶拌样,柱层析纯化分离制得化合物1-1(洗脱剂:石油醚-乙酸乙酯(10∶1,V/V))。
香豆素类荧光衍生试剂L1的合成:将中间体化合物1-1(1.45 g,10 mmol)溶于20 mL无水乙醇,添加10 mL 10% NaOH溶液,95 ℃回流搅拌反应2 h,TLC监测反应进程。待反应结束,溶液冷却至室温,加盐酸调节溶液pH值至2,0 ℃下冷却结晶,生成大量沉淀;静置、抽滤,冰水冲洗;真空干燥,得到目标产物L1。经检测产物纯度高,无需纯化,可直接用于后续反应。
以ACN为溶剂,配制不同浓度的荧光衍生试剂L1及其与氟虫腈衍生化产物SF1的储备液,采用荧光分光光度计扫描检测其荧光光谱性质。荧光分光光度计的狭缝设置为10 nm/10 nm,扫描所用石英皿为1 cm×1 cm,以最大吸收波长进行扫描获取发射波长;同时,根据所测定的发射波长与激发波长计算Stokes位移(发射波长与激发波长之间的差值)。
氟虫腈标准溶液的配制:准确称取0.043 7 g氟虫腈,用ACN溶解并定容至10 mL,配制成1.0×10-2 mol/L的氟虫腈标准溶液,置于4 ℃冰箱中贮存,备用。
DCC溶液的配制:准确称取0.103 2 g DCC,用ACN溶解并定容至5 mL,配制成0.1 mol/L的DCC溶液,置于4 ℃冰箱中贮存,备用。
EDC溶液的配制:准确称取0.095 8 g EDC,用ACN溶解并定容至5 mL,配制成0.1 mol/L的EDC溶液,置于4 ℃冰箱中贮存,备用。
PyBOP溶液的配制:准确称取0.260 2 g PyBOP,用ACN溶解并定容至5 mL,配制成0.1 mol/L的PyBOP溶液,置于4 ℃冰箱中贮存,备用。
DMAP溶液的配制:准确称取0.122 2 g DMAP,用ACN溶解并定容至5 mL,配制成0.2 mol/L的DMAP溶液,置于4 ℃冰箱中贮存,备用。
荧光衍生试剂L1溶液的配制:准确称取0.032 6 g所合成的L1,用ACN溶解并定容至25 mL(利用超声完全促溶),配制成5.0×10-3 mol/L的L1溶液,置于4 ℃冰箱中贮存,备用。
L1与氟虫腈的衍生化反应如图2所示。具体的衍生化产物SF1的制备方法如下:将荧光衍生试剂L1(0.47 g,1 mmoL)和40 mL二氯甲烷加入100 mL茄形瓶中,35 ℃搅拌下依次加入DMAP(0.19 g,0.85 mmoL)、EDC(0.19 g,0.5 mmoL)和氟虫腈(0.32 g,0.4 mmoL),继续反应,TLC监测反应进程。待反应结束后,加入40 mL饱和NaOH溶液和20 mL水(超纯水,以下同),以二氯甲烷萃取(3×50 mL),合并有机相,无水硫酸钠干燥,脱溶,以硅胶柱层析进行分离纯化(洗脱剂为正己烷-乙酸乙酯,3∶1,V/V),得到黄绿色的衍生化产物SF1。
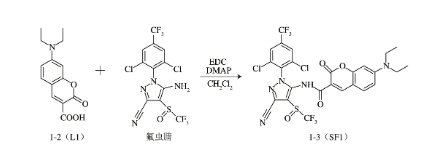
图2 L1与氟虫腈的衍生化反应
取2 mL安瓿瓶,依次加入300 µL DCM、90 µL EDC(0.1 mol/L)、90 µL DMAP(0.2 mol/L)、30 µL氟虫腈溶液(1.0×10-2 mol/L)和660 µL荧光衍生试剂L1(5.0×10-3 mol/L),密封;充分混匀后置于45 ℃水浴中振荡反应60 min,取出冷却至室温,ACN定容至10 mL,取10 µL进行HPLC-FLD检测。
合成的中间体化合物1-1、荧光衍生试剂L1及氟虫腈柱前衍生化产物SF1,均以氢核磁共振(hydrogen nuclear magnetic resonance,1H NMR)、13C核磁共振(13C nuclear magnetic resonance,13C NMR)和高分辨率质谱仪(high resolution mass spectrometer,HRMS)波谱技术进行结构鉴定与表征。
采用单因素试验对衍生化反应主要条件进行优化,试验因素梯度设计如下:催化剂分别选取EDC/DMAP、DCC/DMAP和PyBOP/TEA;反应溶剂分别选取DCM、ACN、THF、DMF、DMSO;反应时间分别设置10、20、30、40、50、60、70、80 min;反应温度分别设置5、15、25、35、45、55、65 ℃;荧光衍生试剂与氟虫腈体积比分别设置1∶1、3∶1、5∶1、7∶1、9∶1、11∶1、13∶1、14∶1。优化试验中的衍生产物,均以HPLC-FLD进行检测,以测得峰面积为指标衡量反应的效率。
HPLC-FLD检测方法和条件如下:Symmetry C18色谱柱(4.6 mm×250 mm,5 μm);柱温40 ℃;进样量10 µL;流速1.0 mL/min;流动相A为100%纯ACN,流动相B为超纯水;等度洗脱,时间为15 min,A相为80%,B相为20%;激发光波长和发射光波长分别为454 nm和490 nm;检测前色谱柱先用流动相平衡10 min,所有检测样品进样前均先经0.22 µm滤膜过滤。
精确吸取优化条件下反应后的溶液10 µL,按照1.3.6节色谱分离条件,连续进样检测20 次,测定衍生化产物SF1的保留时间和峰面积,并计算其相对标准偏差(relative standard deviation,RSD)[33-34]。
将衍生化反应产物在室温条件下分别放置1、2、4、8、24 h后进行HPLC-FLD检测,记录放置不同时间后衍生产物SF1的保留时间和峰面积,并计算其RSD,以此评价检测方法的稳定性[33]。
按照1.3.3.3节氟虫腈柱前衍生化反应条件,进行10 次重复实验,在相同条件下进行HPLC-FLD检测,计算衍生产物SF1的保留时间和峰面积的RSD,以评价方法的重复性[35]。
检出限和定量限分别以RSN=3和RSN=10进行计算,按式(1)计算:

式中:D为检出限;S为检测器灵敏度;N为噪音。
灵敏度S按式(2)计算:

式中:S为灵敏度;I为信号响应值;Q为进样量。
合并2 个公式,得式(3):

信号响应值与噪音之比为该进样量下的信噪比;HPLC-FLD检测时,仪器可自动分析显示信噪比,直接用于计算检出限、定量限。
数据统计分析及绘图软件为Microsoft Excel 2019、IBM SPSS statistics 25、GraphPad Prism 8.0和ChemBioDraw 14。样本间的多重比较采用Duncan新复极差法(P<0.05和P<0.01)。
相关链接:氟虫腈,香豆素,4-(二乙氨基)水杨醛,丙二酸二乙酯,二氯甲烷,二甲基亚砜,北纳生物
本文章来源于——《食品科学网》,如有版权问题,请与本网联系



本期北纳生物将为大家推荐NIM-RM3125 全自动生化分析仪线性范围标准物质。主要用于全自动生化分析仪线性范围项目的校准和检测,该标准物质的均匀性、稳定性良好,质量保障,欢迎大家选购。
了解更多> >
BNCC最新研制的液体室内质控品、核酸标准品火热来袭,本系列标准品通过荧光定量PCR结合数字PCR定值,可用于方法建立、工作标准赋值、方法确认与评价、产品质量控制等。欢迎广大客户朋友们咨询订购!
了解更多> >通话对您免费,请放心接听
温馨提示:
1.手机直接输入,座机前请加区号 如13164239859,010-58103778
2.我们将根据您提供的电话号码,立即回电,请注意接听
3.因为您是被叫方,通话对您免费,请放心接听



登录后才可以评论